



According to Part I of Annex I of Directive 2001/83/EC, a biological medicinal product is a product that contains a biological substance. A biological substance is a substance that is produced by or extracted from a biological source and that needs a combination of physico-chemical-biological testing together with the production process and its control for its characterisation and the determination of its quality. For example, recombinant proteins, monoclonal antibodies, medicinal products derived from human blood and human plasma, immunological medicinal products and advanced therapy medicinal products should be considered biological medicinal products.1 Europe has over 30 years’ experience with biologic medicines.2 By 2014, over 245 biologic medicines had been authorised in the EU and US, representing 166 different active substances.3 A biosimilar medicine is a biologic medicine that contains a version of the active substance of an already authorised biological medicinal product (reference medicine) and which is marketed following the expiry date of exclusivity rights on the reference medicine. The active substance is of biological origin, i.e. it is made by or derived from a biological source, such as a bacterium or yeast. To be authorised, a biosimilar medicine applicant needs to demonstrate that the same clinical benefits can be expected from the biosimilar medicine and its reference medicine based on the fact that all key characteristics for quality, safety and efficacy profiles are the same. The European Medicines Agency (EMA) assesses the scientific data available and issues an opinion on the suitability of a medicine for authorisation for use by patients in the EU. The European Commission decides, based on that opinion, whether or not to authorise a medicine. Beyond the initial authorisation and as with all medicines, the EMA continues to monitor the safety of biosimilar medicines as long as they are in use. These medicines help treat or prevent many diseases ranging from hormone deficiencies, anaemia associated with chronic kidney disease to other widespread and difficult-to-treat diseases such as cancers and auto-immune diseases, rheumatoid arthritis, multiple sclerosis and inflammatory bowel disease. There is already over 30 years of experience with biologic medicines in Europe: the first approved substance for therapeutic use was biosynthetic “human” insulin, first marketed in 1982.4 We now have 10 years of additional experience with biosimilar medicines. Since 2006, EU-approved biosimilar medicines have already generated more than 400 million patient days of clinical experience worldwide. Biosimilar medicines are therefore transforming healthcare in key therapeutic areas today, enhancing safe patient access and the sustainability of European healthcare systems, as demonstrated by the cumulated Real World Evidence (see Glossary below). 1EMA website, accessed March 2016 http://www.ema.europa.eu/ema/index.jsp?curl=pages/regulation/q_and_a/q_and_a_detail_000125.jsp Biologic medicine: a biologic medicine is made up of proteins and is far more complex than well-known chemically synthesised medicines. It is made using living organisms. Biosimilar medicine: a biosimilar medicine is a biologic medicine which is highly similar to the already approved reference product. These medicines treat diseases ranging from hormone deficiencies, anaemia associated with chronic kidney disease to other widespread and difficult-to-treat diseases such as cancers and auto-immune diseases. Biosimilar medicines, like all biologic medicines, are made up of proteins and are far more complex than well-known chemically synthesised medicines and are made using living organisms. Biosimilarity: property of a medicine to show similarity and lack of significant differences in terms of quality, efficacy and safety to a reference biologic medicine to which it has been compared. Extrapolation of indications: extrapolation is a concept allowing a regulatory authority to extend the available data supporting the safety and efficacy of a medicine from one medical condition, disease or disorder (an indication) to another medical condition, disease or disorder (another indication). It is an established scientific and regulatory process which has been used repeatedly for all sorts of medicines and situations. For examples, please refer to “Biosimilars: the science of extrapolation of indication”, Weise M et al. Published in Blood 2014;124(22):3191-3196 – Full text available here (open access). Interchangeability: the medical practice of changing one medicine for another that is expected to achieve the same clinical effect in a given clinical setting and in any patient on the initiative, or with the agreement of, the prescriber.5 Real World Evidence: to demonstrate the clinical and economic value (effectiveness) of an intervention within the healthcare system (e.g. introduction of a new therapeutic option), the data associated with this intervention in the healthcare system can be collated and analysed to provide a unique insight. Such collated data set are called ‘Real World Evidence’ in reference to the fact that they correspond to actual and practical information as opposed to predicted, expected or forecasted value. Reference medicinal product: a medicinal product which has been granted a marketing authorisation by a Member State or by the European Commission on the basis of submitted quality, pre-clinical and clinical data, to which the application for marketing authorisation for a generic or a biosimilar product refers.5 Switching: decision by the treating physician to exchange one medicine for another medicine with the same therapeutic intent in patients who are undergoing treatment.5 5EC Consensus Paper “What you need to know about biosimilar medicinal products”: http://ec.europa.eu/DocsRoom/documents/8242
Definitions
Biologic medicines
Biosimilar medicines
2http://ec.europa.eu/health/human-use/50years/docs/50years_pharma_timeline_v3.pdf
3Biopharmaceutical benchmarks 2014, G. Walsh, Volume 32, Number 10, October 2014, Nature Biotechnology
4European Commission Consensus paper – What you need to know about biosimilar medicines: http://ec.europa.eu/DocsRoom/documents/8242
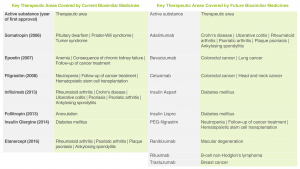
Therapeutic Areas
Glossary
Q&A
Did You Know?